Posologie :1
60 mg/kg de poids corporel,
1 fois par semaine
Contenu d'une boîte :1
1 flacon de poudre 1 g de Repreeza
1 flacon de solvant 20 ml
1 dispositif de transfert avec évent
Indications :1
RESPREEZA est indiqué pour le traitement d'entretien du déficit sévère et documenté en alpha-1 antitrypsine chez les adultes afin de ralentir la progression de l'emphysème (par ex., génotypes PiZZ, PiZ(null), Pi(null,null), PiSZ).
Les patients doivent recevoir un traitement pharmacologique et non pharmacologique optimal et montrer des signes de maladie pulmonaire évolutive (ex : diminution du volume expiratoire maximal par seconde [VEMS] attendu, réduction de la capacité de marche ou augmentation du nombre d'exacerbations) évalués par un professionnel de santé expérimenté dans le traitement du déficit en alpha-1 antitrypsine.
▼ Ce médicament fait l'objet d'une surveillance supplémentaire qui permettra l'identification rapide de nouvelles informations relatives à la sécurité. Les professionnels de la santé déclarent tout effet indésirable suspecté. Voir rubrique 4.8 pour les modalités de déclaration des effets indésirables.
Sélection des donneurs et sécurisation du plasma6
Respreeza est une spécialité dérivée du plasma humain. CSL Behring possède CSL Plasma, l’un des plus grands réseaux de collecte de plasma au monde, qui nous permet de garantir chaque jour aux patients l’accès à leurs traitements.
L’origine du plasma ainsi collecté est parfaitement identifiée : les donneurs, non-rémunérés, sont strictement sélectionnés afin d’exclure les donneurs à risque.
Les plasmas collectés sont ensuite mis en quarantaine et testés biologiquement. Cette qualification biologique permet de valider la qualité et la sécurité du plasma.
Des étapes spécifiques de sécurisation sont intégrées au processus de fractionnement. Elles ont pour objectif de réduire le risque viral et prion sans dénaturer les protéines plasmatiques, généralement plus fragiles.
L’efficacité des mesures mises en place est testée dans une unité dédiée soit sur des modèles de virus validés (ex : le parvovirus canin est le modèle du parvovirus B19) , soit sur virus réel (VIH-1, virus de l’hépatite A…). La réduction est jugée efficace au-delà d’un facteur supérieur à 4 log 10.
Les mesures mises en place pour Respreeza sont considérées comme efficaces vis-à-vis des virus enveloppés tels que le VIH, le VHB et le VHC et pour les virus non-enveloppés du VHA et du parvovirus B191.
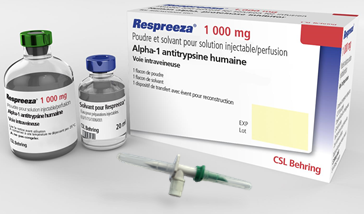
Schéma simplifié du processus de fabrication de Respreeza4,5
Recommandations EMA7
Il est généralement préférable d’associer 2 étapes spécifiques, complémentaires par leur mode d’action, de telle manière qu’un virus survivant à la première étape soit efficacement réduit/éliminé par la seconde ; au moins l’une des étapes devrait être efficace contre les virus non-enveloppés.
Méthode de Cohn modifiée4
Cette méthode de fractionnement repose sur une technique de cryoprécipitation suivie de précipitations par l’alcool successives.
Cette étape contribue à la sécurisation vis-à-vis du risque viral et prion.
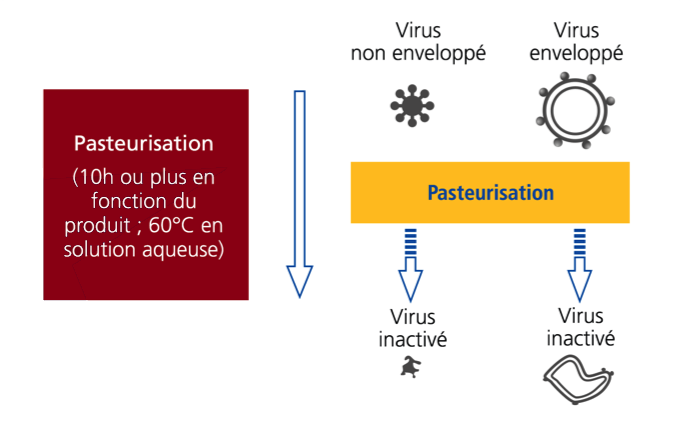
Pasteurisation4
La solution est incubée au moins 10h à 60°C.
Cette étape est efficace contre les virus enveloppés et non-enveloppés.
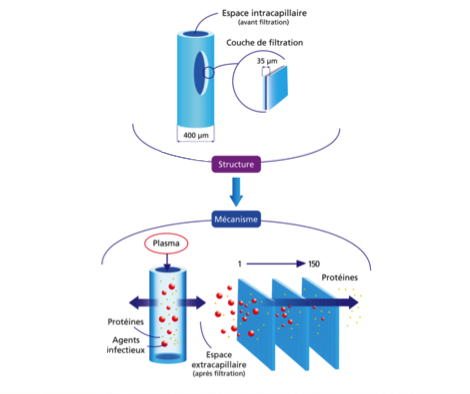
Nanofiltration 20 nm4
Elle permet d’éliminer les particules virales de taille supérieure au diamètre du filtre. Elle élimine tous les types d’agents infectieux, sans dénaturer les protéines ni affecter la qualité du produit.
Cette étape est efficace contre les virus enveloppés et non-enveloppés.
Agents infectieux transmissibles1
Les mesures habituelles de prévention du risque de transmission d’infections par les médicaments préparés à partir de sang ou de plasma humain comprennent la sélection des donneurs, la recherche des marqueurs spécifiques d’infection sur chaque don et sur les mélanges de plasma ainsi que la mise en œuvre dans le procédé de fabrication d’étapes efficaces pour l’inactivation/élimination virale.
Cependant, lorsque des médicaments préparés à partir de sang ou de plasma humain sont administrés, le risque de transmission d’agents infectieux ne peut pas être totalement exclu.
Ceci s’applique également aux virus inconnus ou émergents ou d’autres types d’agents infectieux. Les mesures mises en place sont considérées comme efficaces vis-à-vis des virus enveloppés tels que le virus de l’immunodéficience humaine (VIH) et les virus de l’hépatite B (VHB) et de l’hépatite C (VHC) et pour les virus non enveloppés de l’hépatite A (VHA) et du parvovirus B19.
Une vaccination appropriée (hépatite A et B) des patients recevant régulièrement ou de façon répétée des spécialités d’alpha-1 antitrypsine préparées à partir de plasma humain est recommandée.
Solution concentrée à 5%1
-
50 mg d’alpha-1 antitrypsine humaine par ml
Moins de volume à perfuser à chaque cure*
- Exemple pour un patient de 70 kg :
- Dose hebdomadaire : 4g de Respreeza
- Volume à administrer : 80 ml
Propriétés pharmaceutiques1
-
Demi-vie : 5,1 (+ 2,4) jours
A1AT endogène : 4,5 jours
-
pH : 7,0
plasma sanguin : 7,35 – 7,45
-
Osmolarité : 279 mOsmol/kg
plasma sanguin : 280 à 300 mOsmol/kg
-
Sodium : 1,9 mg/ml
soit 38 mg/g de Respreeza
Solution purifiée8
> 99% de pureté (déterminé par HPLC)
< 0,001 mg d’IgA par mg de protéine (non-détectable par HPLC)
Posologie
60 mg/kg de poids corporel,
1 fois par semaine
Administration
Débit recommandé : 0,08 ml/kg de p.c./min
Durée de perfusion ≈ 15 minutes (à la posologie recommandée)
Conservation
3 ans à une température ne dépassant pas 25°C
3 heures après reconstitution à température ambiante (≤ 25°C)
Ne pas congeler
-
Exemple pour un patient de 70 kg :
- Dose hebdomadaire : 4g de Respreeza
- Volume à administrer : 80 ml
- Durée d'administration : 15 min
Documents à votre disposition
Livret de suivi de traitement Respreeza
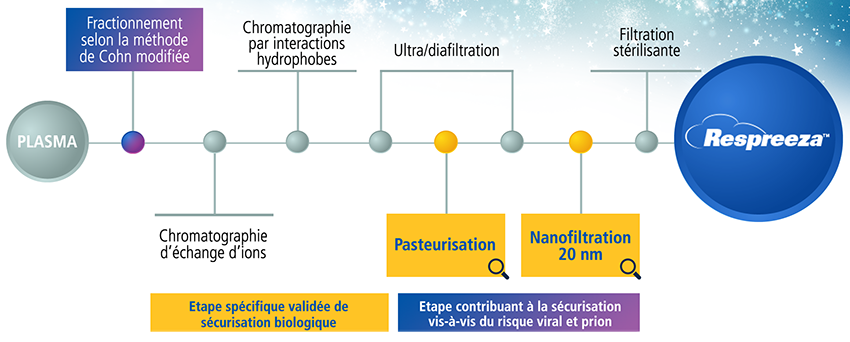
Méthodologie9
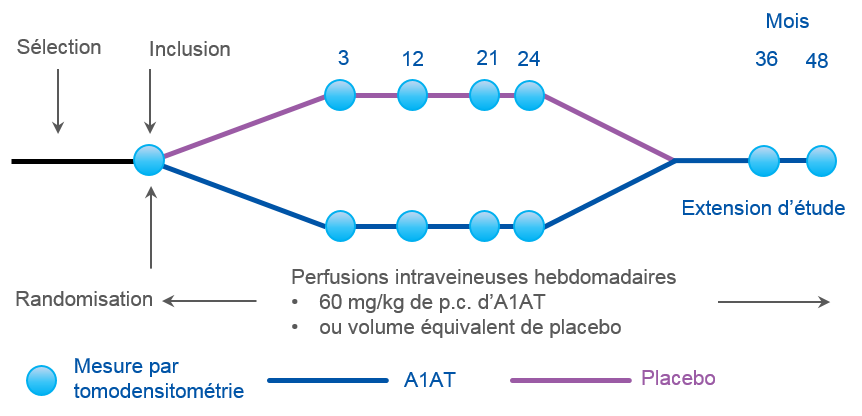
Etude multicentrique (28 centres dans 13 pays) de phase III/IV de supériorité, prospective, randomisée, comparative vs placebo, en double-aveugle.
L’objectif est d’évaluer l’effet de Respreeza sur la progression de l’emphysème, évaluée par le déclin de la densité parenchymateuse pulmonaire et mesurée par tomodensitométrie.
Principaux critères d’inclusion : Patients âgés de 18 à 65 ans ayant un déficit en alpha-1 antitrypsine (taux sérique < 11 µM), un emphysème et un VEMS compris entre 35 % et 70 % de la valeur théorique.
Principaux critères de non-inclusion : Tabagisme actif, déficit en IgA, transplantation pulmonaire ou chirurgie de réduction pulmonaire.
Population ITT : 180 patients randomisés en 2 groupes : 93 sujets dans le groupe Respreeza et 87 sujets dans le groupe Placebo.
Traitement : alpha-1 antitrypsine humaine en perfusion IV, 60 mg/kg/semaine pendant 24 mois.
Critère de jugement principal : « Adjusted P15 » : Variation annuelle de la densité parenchymateuse pulmonaire ajustée au volume pulmonaire (g/L) (estimée au 15eme percentile), mesurée par tomodensitométrie réalisée à 2 temps respiratoires différents : en inspiration forcée (CPT : Capacité Pulmonaire Totale) et en expiration (CRF : Capacité Résiduelle Fonctionnelle), à l’inclusion, puis à 3, 12, 21 et 24 mois.
Schéma de l’étude RAPID9
Efficacité9
La majorité des patients inclus dans l’étude était de phénotype PIZZ (92,2 %).
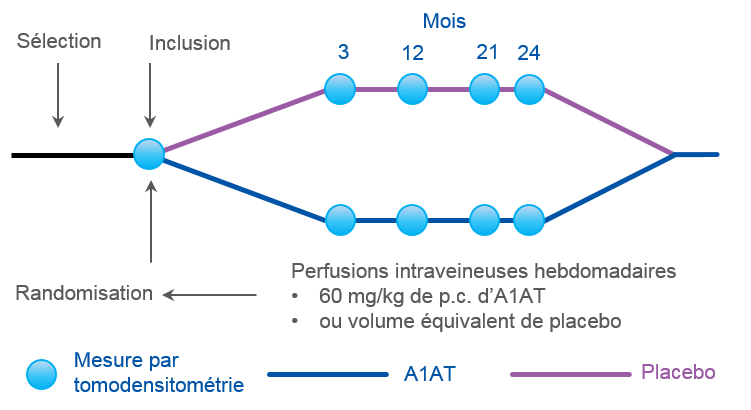
Sur le critère principal d’évaluation, en inspiration maximale (CPT), un ralentissement d’environ 34 % de la progression de l’emphysème était mis en évidence dans le groupe Respreeza par rapport au groupe placebo (p=0,017).
La diminution de la densité parenchymateuse pulmonaire après 24 mois était d’environ 2,6 g/L dans le groupe Respreeza, vs environ 4,2 g/L dans le groupe placebo.
La mesure en expiration (CRF) et le critère combiné (moyenne des 2 temps respiratoires) n’ont pas mis en évidence de différence statistiquement significative de Respreeza par rapport au placebo même s’il existe une tendance positive en faveur de Respreeza.
Respreeza permet donc de réduire significativement le taux annuel de déclin de la densité pulmonaire mesurée au scanner en capacité pulmonaire totale chez les patients en déficit de A1AT.
Variation annuelle de la densité parenchymateuse pulmonaire (population ITT)
| Stade respiratoire | Variation annuelle de la densité pulmonaire (Adj P15) en g/L, Moyenne (écart-type) | Différence RESPREZA vs Placebo | |||
|---|---|---|---|---|---|
| Respreeza | Placebo | Différence | IC 95% | p* | |
| Capacité pulmonaire totale | -1,45 (0,23) | -2,19 (0,25) | 0,74 | [0,06-1,42] | 0,017 |
| Capacité résiduelle fonctionnelle | -1,55 (0,24) | -2,02 (0,26) | 0,48 | [-0,22-1,18] | 0,090 |
| Mesure combinée en CPT + CRF | -1,50 (0,22) | -2,12 (0,24) | 0,62 | [-0,22-1,26] | 0,029 |
|
*Analyse unilatérale avec un risque α = 2,5 % |
|||||

Sur les critères secondaires d’évaluation, Il n’a pas été mis en évidence de différence significative entre les deux groupes.
Tolérance9
Le profil de tolérance de RESPREEZA était similaire à celui du placebo en terme de nombre de patients ayant eu un événement indésirable grave et de la fréquence d’événements indésirables graves.
L’événement indésirable le plus fréquemment rapporté et considéré en lien avec le traitement a été une céphalée (10,8 % des patients du bras RESPREEZA et 5,7 % patients du bras placebo).
Dans le groupe RESPREEZA, aucun événement indésirable grave considéré comme relié au produit n’est survenu au cours des 24 heures suivant l’administration.
Evénements indésirables rapportés dans l’étude RAPID (24 mois de suivi)
| Respreeza (n=93) | Placebo (n=87) | |||
|---|---|---|---|---|
| Evénements (n) | Incidence Taux | Evénements (n) | Incidence Taux | |
| Tout effet indesirable (AE) | 1298 (92) | 7,58 | 1068 (86) | 7,23 |
| Tout effet indésirable d'une durée < 24h | 373 (78) | 2,18 | 328 (78) | 2,22 |
| Toute réaction secondaire | 702 (86) | 4,10 | 560 (83) | 3,79 |
| Tout effet indésirable lié au trt | 91 (21) | 0,53 | 50 (21) | 0,34 |
Quatre décès, dont aucun n’a été considéré comme relié au traitement, sont survenus au cours de l’étude RAPID : 1 décès (insuffisance respiratoire) dans le bras RESPREEZA et 3 décès (sepsis, pneumonie et cancer du sein métastatique) dans le bras placebo.
Pour l’ensemble des effets indésirables de Respreeza, se référer à la rubrique « Bon Usage » et aux mentions légales.
Méthodologie9
Etude d’extension en ouvert de l’étude RAPID.
Son objectif est d’évaluer l’efficacité de RESPREEZA à long terme sur la variation de la densité parenchymateuse pulmonaire chez les patients déficitaires en alpha-1 antitrypsine présentant un emphysème pulmonaire ayant terminé l’étude RAPID.
Principal critère d’inclusion : Patients ayant participé à l’étude RAPID.
Principal critère d’exclusion : Participer dans un centre aux Etats-Unis.
Population ITT : 140 patients répartis en 2 groupes : 76 patients dans le groupe « instauration immédiate » et 64 patients dans le groupe
« instauration retardée ».
Traitement : alpha-1 antitrypsine humaine en perfusion IV, 60 mg/kg/semaine pendant 24 mois.
Critère de jugement principal : « Adjusted P15 » : Variation annuelle de la densité parenchymateuse pulmonaire ajustée au volume pulmonaire (g/L) (estimée au 15eme percentile), mesurée par tomodensitométrie réalisée à 2 temps respiratoires différents : en inspiration forcée (CPT) et en expiration (CRF), à l’inclusion, puis à 3, 12, 21 et 24 mois.
Schéma de l’étude RAPID incluant la phase d’extension9
Efficacité9
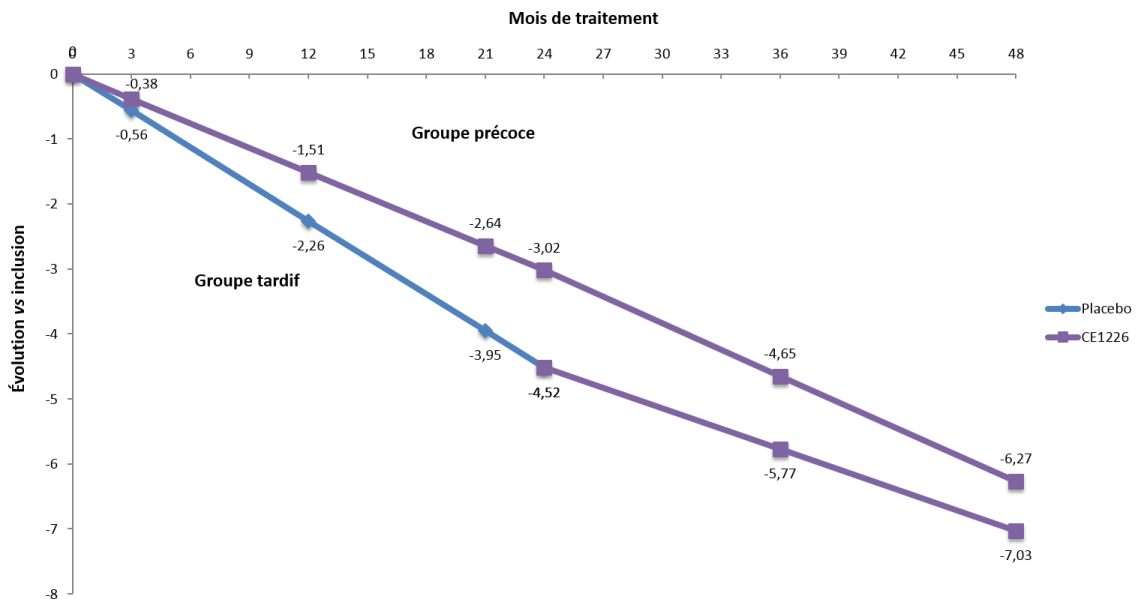
Dans le groupe « Instauration immédiate », déjà traité par Respreeza dans l’étude RAPID, les variations de perte de densité pulmonaire ont été du même ordre dans RAPID et dans la phase d’extension.
Dans le groupe RESPREEZA « Instauration retardée », une différence a été observée entre la période où les patients ont reçu le placebo et la période de suivi en ouvert pendant laquelle ils ont reçu RESPREEZA (-2,3 g/L/an dans RAPID et -1,3 g/L/an dans la phase d’extension).
Variation annuelle de densité pulmonaire (AdjP15)
| Variation annuelle de la densité pulmonaire en g/L/an, moyenne (écart-type) |
||
|---|---|---|
| Δ entre l'inclusion et 24 mois (étude RAPID) |
Δ entre 24 et 48 mois (étude RAPID Extension) |
|
| Population ITT | ||
| Groupe "Instauration immédiate", N=75 | -1,509 (0,2483) | -1,627 (0,2743) |
| Groupe "Instauration retardée", N=64 | -2,259 (0,2679) | -1,256 (0,2891) |
| Population Completer* | ||
| Groupe "Instauration immédiate", N=62 | -1,595 (0,2730) | -1,435 (0,2655) |
| Groupe "Instauration immédiate", N=57 | -2,347 (0,2836) | -1,265 (0,2764) |
| *Population Completer : population comprenant les patients avec une valeur initiale de densité pulmonaire (Jour 1 de l’étude RAPID) et une valeur au mois 48 de l’étude d’extension | ||
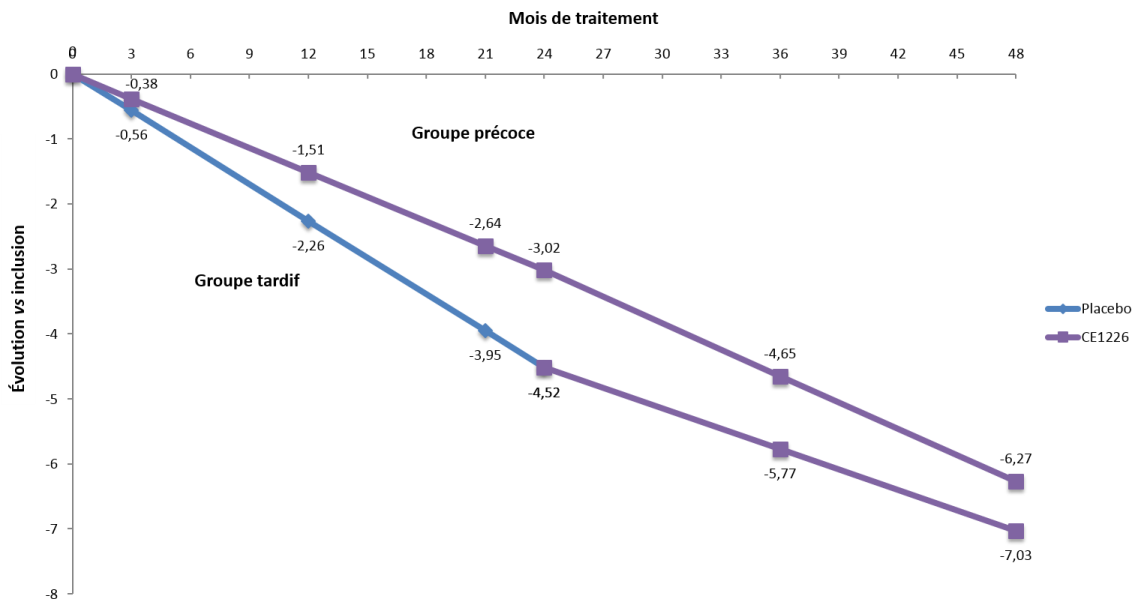
Le ralentissement de la perte de densité pulmonaire sous RESPREEZA chez les patients du groupe« Instauration retardée » (-1,3 g/L/an) était similaire à celui observé chez les patients traités par RESPREEZA au cours de l’étude RAPID (-1,5 g/L/an) et de la phase d’extension (-1,6 g/L /an).
La phase d’extension sur 24 mois a donc mis en évidence :
- Un maintien de l’efficacité du traitement par RESPREEZA sur la progression de l’emphysème durant les 4 années de traitement,
- Un ralentissement de la perte de densité pulmonaire chez les patients du groupe placebo traités par RESPREEZA lors de l’inclusion dans la phase d’extension comparable à celui observé dans le groupe traité par RESPREEZA dans l’étude RAPID.
Tolérance9
L’ensemble des patients inclus a été traité par RESPREEZA.
Parmi les 140 patients, il a été rapporté un événement indésirable chez 138 (98,6%) patients : tous les patients du groupe « Instauration immédiate »
et 96,9% des patients du groupe « Instauration retardée ».
Evénements indésirables (EI) rapportés au cours de la phase d’extension (24 mois)
| Groupe Initiation immédiate (N=76) | Groupe Initiation retardée (N=64) | Total (N=140) | ||||
|---|---|---|---|---|---|---|
| Nb de patients (%) | Nb d’événements (fréquence) | Nb de patients (%) | Nb d’événements (fréquence) | Nb de patients (%) | Nb d’événements (fréquence) | |
| EI | 76 (100,0) | 773 (5,28) | 62 (96,9) | 620 (4,97) | 138 (98,6) | 1393 (5,14) |
| EI considéré comme relié au traitement | 11 (14,5) | 21 (0,14) | 7 (10,9) | 7 (0,06) | 18 (12,9) | 28 (0,10) |
| EI survenu dans les 24h suivant l’administration | 66 (86,8) | 250 (1,71) | 51 (79,7) | 173 (1,39) | 117 (83,6) | 423 (1,56) |
| EI considéré comme relié au traitement survenu dans les 24h suivant l’administration | 5 (6,6) | - | 5 (7,8) | - | 10 (7,1) | - |
| EI grave | 28 (36,8) | 57 (0,39) | 23 (35,9) | 56 (0,45) | 51 (36,4) | 113 (0,42) |
| EI grave considéré comme relié au traitement | 1 (1,3) | 1 (0,01) | 3 (4,7) | 3 (0,02) | 4 (2,9) | 4 (0,01) |
| EI grave ayant conduit à la sortie de l’étude | 1 (1,3) | 1 (0,01) | 1 (1,6) | 1 (0,01) | 2 (1,4) | 2 (0,01) |
| Décès lié à un EI | 1 (1,3) | 1 (0,01) | 0 | 0 | 1 (0,7) | 1 (0,00) |
| Décès lié à un EI considéré comme relié au traitement | 0 | 1 (0,01) | 0 | 0 | 0 | 0 |
Les événements indésirables les plus fréquemment rapportés ont été : BPCO (bronchopneumopathie chronique obstructive), infection des voies respiratoires inférieures, aggravation de la pathologie, nasopharyngite et céphalée.
Un décès consécutif à un événement indésirable considéré comme non relié au traitement (exacerbation de la BPCO du patient) est survenu au cours de la phase d’extension.
Pour l’ensemble des effets indésirables de Respreeza, se référer à la rubrique « Bon Usage » et aux mentions légales.
Effets indésirables
Résumé du profil de sécurité
Des réactions allergiques ou d'hypersensibilité ont été observées pendant le traitement. Dans les cas les plus graves, les réactions allergiques peuvent évoluer vers des réactions anaphylactiques sévères, même lorsque le patient n'a pas présenté d'hypersensibilité à des administrations antérieures (voir « Mises en garde spéciales et précautions d’emploi »).
Tableau des effets indésirables
Mises en garde spéciales et précautions d’emploi
La vitesse de perfusion recommandée à la rubrique 4.2 doit être respectée. L'état clinique du patient (y compris les signes vitaux) doit être étroitement surveillé tout au long des premières perfusions. Si des réactions potentiellement liées à l’administration de Respreeza apparaissent, la perfusion doit être ralentie ou arrêtée, en fonction de l’état clinique du patient. Si les symptômes s'estompent rapidement après l'arrêt de la perfusion, celle-ci peut être reprise à une vitesse plus lente ne compromettant pas le confort du patient.
Hypersensibilité : Des réactions d'hypersensibilité peuvent survenir, y compris chez les patients ayant toléré un traitement précédent par alpha-1 antitrypsine humaine. En fonction de leur nature et de leur sévérité, les réactions de type allergique ou anaphylactique suspectées peuvent exiger l'arrêt immédiat de la perfusion En cas de choc, un traitement médical d'urgence doit être administré.
Traitement à domicile/auto-administration : Les données sur l'utilisation de ce médicament à domicile/en auto-administration sont limitées. Les risques potentiels associés au traitement à domicile/à l'auto-administration sont liés à la manipulation et à l'administration du médicament ainsi qu'à la prise en charge des réactions indésirables, en particulier l'hypersensibilité. Les patients doivent être informés des signes des réactions d'hypersensibilité. Le bien-fondé du traitement à domicile/de l'auto-administration pour le patient est laissé à l'appréciation du médecin traitant qui doit s'assurer qu'une formation appropriée est fournie (ex. : concernant la reconstitution, l'utilisation du dispositif de transfert ou du filtre, l'assemblage de la tubulure de perfusion intraveineuse, les techniques de perfusion, la tenue d'un livret de traitement, l'identification des effets indésirables et les mesures à prendre si un tel effet se manifeste) et que le traitement est réévalué à intervalles réguliers.
Agents infectieux transmissibles : Les mesures habituelles de prévention du risque de transmission d’infections par les médicaments
préparés à partir de sang ou de plasma humain comprennent la sélection des donneurs, la recherche des marqueurs spécifiques d’infection sur chaque don et sur les mélanges de
plasma ainsi que la mise en œuvre dans le procédé de fabrication d’étapes efficaces pour l’inactivation/élimination virale.
Cependant, lorsque des médicaments préparés à partir de sang ou de plasma humain sont administrés, le risque de transmission d’agents infectieux ne peut pas être totalement exclu.
Ceci s’applique également aux virus inconnus ou émergents ou d’autres types d’agents infectieux.
Les mesures mises en place sont considérées comme efficaces vis-à-vis des virus enveloppés tels que le virus de l’immunodéficience humaine (VIH) et
les virus de l’hépatite B (VHB) et de l’hépatite C (VHC) et pour les virus non enveloppés de l’hépatite A (VHA) et du parvovirus B19.
Une vaccination appropriée (hépatite A et B) des patients recevant régulièrement ou de façon répétée des spécialités d’alpha-1 antitrypsine préparées à partir de
plasma humain est recommandée.
Tabagisme : La fumée de tabac est un facteur de risque important d'apparition et de progression de l'emphysème. Il est donc fortement recommandé d'arrêter de fumer et d'éviter la fumée de tabac environnementale.
Contenu en sodium : Respreeza contient environ 1,9 mg (< 1 mmol) de sodium par ml de solution reconstituée. ceci doit être pris en compte pour les patients suivant un régime contrôlé en sodium.
Contre-indications
Hypersensibilité au principe actif ou à l’un des excipients (chlorure de sodium, phosphate monosodique monohydraté, mannitol) (voir également « Mises en garde spéciales et précautions d’emploi »).
Patients souffrant d'un déficit en IgA chez qui la présence d'anticorps anti-IgA a été démontrée, en raison du risque de réactions anaphylactiques et d'hypersensibilité sévères.
Interactions avec d’autres médicaments et autres formes d’interactions
Aucune étude d’interaction n’a été réalisée.
Conditions de prescription et de délivrance
Liste I. Agréé aux collectivités.10
Médicament soumis à prescription hospitalière. La délivrance est réservée aux pharmacies à usage intérieur des établissements de santé.
Inscrit sur liste de rétrocession avec prise en charge.11
TR RESPREEZA 1000 mg : en cours.
- Mentions légales Respreeza
- Interim Safety Analysis, Page 1 of 8 08Jul03_07Jul13
- Chapman KR, Burdon JGW, Piitulainen E, et al. lntravenous augmentation treatment and lung density in severe α1 antitrypsin deficiency (RAPID): a randomised, double-blind, placebo-controlled trial. Lancet. 2015;386(9991 ):360-368
- Dossier AMM RESPREEZA 3.2.A.2.1 Adventitious Agents Safety Evaluation (pages 1-9) & Attachment 1 (pages 8-9)
- EMA – Respreeza : EPAR- Public Assessment Report 16/09/2015 EMA/CHMP/76739/2015 2.2.2. Active substance
- CSL Behring Plasma Master File 2015, Module 3.1.2 pages 1-5
- EMA – Guideline on plasma-derived medicinal products EMA/CHMP/BWP/706271/2010
- Cowden Dl, Fisher GE, Weeks RL. A pilot study comparing the purity, functionality and isoform composition of alpha-1 -proteinase inhibitor (human) products. Curr Med Res Opin. 2005;21 (6):877-883
- Avis de transparence Respreeza du 3 février 2016
- JO n°60 du 11 mars 2016
- JO n°143 du 21 juin 2016
Principe de la pasteurisation
Principe de la nanofiltration
Evolution de la variation annuelle de densité pulmonaire (Adj P15) en inspiration maximale (population ITT)9
Variation annuelle de la densité parenchymateuse pulmonaire (population ITT)9